Can I Have Familial Hypercholesterolemia if Neither of My Parents Have High Cholesterol
| Familial hypercholesterolemia | |
|---|---|
| Other names | Familial hypercholesterolaemia |
 | |
| Xanthelasma palpebrarum, yellow patches consisting of cholesterol deposits above the eyelids. These are more common in people with FH. | |
| Specialty | Endocrinology |
Familial hypercholesterolemia (FH) is a genetic disorder characterized by loftier cholesterol levels, specifically very high levels of low-density lipoprotein (LDL, "bad cholesterol"), in the blood and early on cardiovascular illness. The most common mutations diminish the number of functional LDL receptors in the liver.[ commendation needed ] Since the underlying torso biochemistry is slightly different in individuals with FH, their high cholesterol levels are less responsive to the kinds of cholesterol control methods which are usually more effective in people without FH (such equally dietary modification and statin tablets). Nevertheless, treatment (including higher statin doses) is usually effective.
FH is classified as a type 2 familial dyslipidemia.[1] At that place are five types of familial dyslipidemia (non including subtypes), and each are classified from both the altered lipid profile and past the genetic aberration. For example, high LDL (often due to LDL receptor defect) is type 2. Others include defects in chylomicron metabolism, triglyceride metabolism, and metabolism of other cholesterol-containing particles, such as VLDL and IDL.
Most 1 in 100 to 200 people accept mutations in the LDLR gene that encodes the LDL receptor poly peptide, which normally removes LDL from the circulation, or apolipoprotein B (ApoB), which is the part of LDL that binds with the receptor; mutations in other genes are rare.[2] People who have one abnormal copy (are heterozygous) of the LDLR gene may develop cardiovascular disease prematurely at the age of 30 to 40. Having two abnormal copies (beingness homozygous) may cause severe cardiovascular disease in babyhood. Heterozygous FH is a common genetic disorder, inherited in an autosomal ascendant design, occurring in ane:250 people in most countries;[iii] homozygous FH is much rarer, occurring in 1 in 300,000 people.[ commendation needed ]
Heterozygous FH is ordinarily treated with statins, bile acid sequestrants, or other lipid-lowering agents that lower cholesterol levels. New cases are by and large offered genetic counseling. Homozygous FH frequently does non respond to medical therapy and may require other treatments, including LDL apheresis (removal of LDL in a method similar to dialysis) and occasionally liver transplantation.[4]
Signs and symptoms [edit]
Physical signs [edit]
High cholesterol levels normally do not cause whatsoever symptoms. Yellow deposits of cholesterol-rich fat may be seen in various places on the body such as around the eyelids (known equally xanthelasma palpebrarum), the outer margin of the iris (known as arcus senilis corneae), and in the tendons of the hands, elbows, knees and feet, particularly the Achilles tendon (known as a tendon xanthoma).[4] [5]
Cardiovascular disease [edit]
Accelerated deposition of cholesterol in the walls of arteries leads to atherosclerosis, the underlying cause of cardiovascular disease. The most common problem in FH is the evolution of coronary artery disease (atherosclerosis of the coronary arteries that supply the heart) at a much younger age than would exist expected in the general population. This may lead to angina pectoris (breast pain or tightness on exertion) or heart attacks. Less unremarkably, arteries of the encephalon are afflicted; this may lead to transient ischemic attacks (brief episodes of weakness on one side of the torso or disability to talk) or occasionally stroke. Peripheral avenue occlusive disease (obstruction of the arteries of the legs) occurs mainly in people with FH who smoke; this tin can cause pain in the calf muscles during walking that resolves with rest (intermittent claudication) and problems due to a decreased claret supply to the feet (such as gangrene).[6] Atherosclerosis hazard is increased further with historic period and in those who smoke, accept diabetes, high blood force per unit area and a family unit history of cardiovascular disease.[4] [7]
Diagnosis [edit]
| Criteria for diagnosis of probable heterozygous FH (98% specificity)[eight] | ||||||
|---|---|---|---|---|---|---|
| 1st degree relative | general population | |||||
| age | cholesterol | mg/dL | mmol/L | mg/dL | mmol/L | |
| < 18 | full | > 220 | > 5.seven | > 270 | > seven.0 | |
| LDL-C | > 155 | > four.0 | > 200 | > 5.ii | ||
| 20–29 | total | > 240 | > half-dozen.2 | > 290 | > 7.5 | |
| LDL-C | > 170 | > four.iv | > 220 | > 5.vii | ||
| xxx–39 | total | > 270 | > 7.0 | > 340 | > eight.8 | |
| LDL-C | > 190 | > five.0 | > 240 | > six.2 | ||
| ≥ 40 | full | > 290 | > 7.5 | > 360 | > ix.3 | |
| LDL-C | > 205 | > 5.iii | > 260 | > 6.vii | ||
| First-caste relatives are parents, offspring, brothers, and sisters | ||||||
Approximately 85% of individuals with this disorder have not been diagnosed and consequently are not receiving lipid-lowering treatments.[ix] Concrete test findings tin can assist a doctor brand the diagnosis of FH. Tendon xanthomas are seen in 20-40% of individuals with FH and are pathognomonic for the condition.[ix] A xanthelasma or corneal arcus may also be seen. These common signs are supportive of the diagnosis, merely are non-specific findings.[9]
Lipid measurements [edit]
Cholesterol levels may be determined equally part of health screening for health insurance or occupational wellness, when the external physical signs such as xanthelasma, xanthoma, arcus are noticed, symptoms of cardiovascular illness develop, or a family member has been found to accept FH. A pattern compatible with hyperlipoproteinemia type IIa on the Fredrickson classification is typically constitute: raised level of full cholesterol, markedly raised level of low-density lipoprotein (LDL), normal level of loftier-density lipoprotein (HDL), and normal level of triglycerides. Total cholesterol levels of 350–550 mg/dL are typical of heterozygous FH while full cholesterol levels of 650–thou mg/dL are typical of homozygous FH.[9] The LDL is typically above the 75th percentile, that is, 75% of the healthy population would accept a lower LDL level.[four] Cholesterol levels can exist drastically higher in people with FH who are too obese.[six]
Mutation analysis [edit]
On the ground of the isolated loftier LDL and clinical criteria (which differ by country), genetic testing for LDL receptor mutations and ApoB mutations tin can be performed. Mutations are detected in between 50 and 80% of cases; those without a mutation often have higher triglyceride levels and may in fact have other causes for their high cholesterol, such every bit combined hyperlipidemia due to metabolic syndrome.[ten]
Differential diagnosis [edit]
FH needs to exist distinguished from familial combined hyperlipidemia and polygenic hypercholesterolemia. Lipid levels and the presence of xanthomata can confirm the diagnosis. Sitosterolemia and cerebrotendineous xanthomatosis are two rare weather condition that tin likewise present with premature atherosclerosis and xanthomas. The latter condition tin can also involve neurological or psychiatric manifestations, cataracts, diarrhea and skeletal abnormalities.[11]
Genetics [edit]
The most common genetic defects in FH are LDLR mutations (prevalence 1 in 250, depending on the population),[three] ApoB mutations (prevalence 1 in 1000), PCSK9 mutations (less than ane in 2500) and LDLRAP1. The related affliction sitosterolemia, which has many similarities with FH and also features cholesterol aggregating in tissues, is due to ABCG5 and ABCG8 mutations.[4]
LDL receptor [edit]


Schematic representation of the LDL receptor protein.
The LDL receptor gene is located on the curt arm of chromosome 19 (19p13.1-xiii.3).[ix] It comprises 18 exons and spans 45 kb, and the protein gene production contains 839 amino acids in mature form. A single aberrant copy (heterozygote) of FH causes cardiovascular disease by the age of 50 in virtually 40% of cases. Having two abnormal copies (homozygote) causes accelerated atherosclerosis in childhood, including its complications. The plasma LDL levels are inversely related to the action of LDL receptor (LDLR). Homozygotes accept LDLR action of less than 2%, while heterozygotes take defective LDL processing with receptor activity being two–25%, depending on the nature of the mutation. Over 1000 different mutations are known.[4]
There are five major classes of FH due to LDLR mutations:[12]
- Form I: LDLR is not synthesized at all.
- Class 2: LDLR is not properly transported from the endoplasmic reticulum to the Golgi apparatus for expression on the cell surface.
- Form Iii: LDLR does non properly bind LDL on the cell surface because of a defect in either apolipoprotein B100 (R3500Q) or in LDL-R.
- Class 4: LDLR bound to LDL does not properly cluster in clathrin-coated pits for receptor-mediated endocytosis (pathway step 2).
- Class V: LDLR is not recycled dorsum to the jail cell surface (pathway step 5).
Apolipoprotein B [edit]
Apolipoprotein B, in its ApoB100 form, is the main apolipoprotein, or protein part of the lipoprotein particle. Its gene is located on the second chromosome (2p24-p23) and is 46.2 kb long. FH is frequently associated with the mutation of R3500Q, which causes replacement of arginine by glutamine at position 3500. The mutation is located on a part of the poly peptide that normally binds with the LDL receptor, and binding is reduced as a effect of the mutation. Like LDLR, the number of aberrant copies determines the severity of the hypercholesterolemia.[4] [13]
PCSK9 [edit]
Mutations in the proprotein convertase subtilisin/kexin type 9 (PCSK9) gene were linked to autosomal dominant (i.e. requiring simply one abnormal re-create) FH in a 2003 report.[4] [14] The factor is located on the first chromosome (1p34.1-p32) and encodes a 666 amino acid poly peptide that is expressed in the liver. It has been suggested that PCSK9 causes FH mainly past reducing the number of LDL receptors on liver cells.[xv]
LDLRAP1 [edit]
Abnormalities in the ARH gene, also known as LDLRAP1, were first reported in a family in 1973.[16] In contrast to the other causes, two abnormal copies of the gene are required for FH to develop (autosomal recessive). The mutations in the protein tend to cause the production of a shortened protein. Its real function is unclear, only it seems to play a office in the relation between the LDL receptor and clathrin-coated pits. People with autosomal recessive hypercholesterolemia tend to accept more severe disease than LDLR-heterozygotes but less severe than LDLR-homozygotes.[iv]
Pathophysiology [edit]
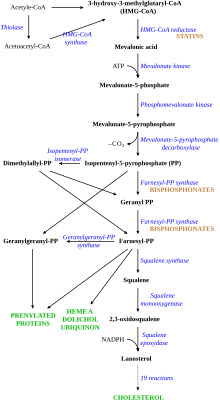
LDL cholesterol normally circulates in the body for two.5 days, and later the apolipoprotein B portion of LDL cholesterol binds to the LDL receptor on the liver cells, triggering its uptake and digestion.[9] This process results in the removal of LDL from the circulatory organisation. Synthesis of cholesterol past the liver is suppressed in the HMG-CoA reductase pathway.[17] In FH, LDL receptor function is reduced or absent-minded,[nine] and LDL circulates for an average duration of iv.5 days, resulting in significantly increased level of LDL cholesterol in the blood with normal levels of other lipoproteins.[6] In mutations of ApoB, reduced binding of LDL particles to the receptor causes the increased level of LDL cholesterol. Information technology is not known how the mutation causes LDL receptor dysfunction in mutations of PCSK9 and ARH.[4]
Although atherosclerosis occurs to a certain degree in all people, people with FH may develop accelerated atherosclerosis due to the excess level of LDL. The degree of atherosclerosis approximately depends on the number of LDL receptors all the same expressed and the functionality of these receptors. In many heterozygous forms of FH, the receptor function is only mildly impaired, and LDL levels will remain relatively low. In the more serious homozygous forms, the receptor is not expressed at all.[four]
Some studies of FH cohorts suggest that additional hazard factors are generally at play when a person develops atherosclerosis.[xviii] [19] In add-on to the classic risk factors such as smoking, high blood force per unit area, and diabetes, genetic studies have shown that a mutual abnormality in the prothrombin gene (G20210A) increases the adventure of cardiovascular events in people with FH.[20] Several studies establish that a high level of lipoprotein(a) was an additional run a risk factor for ischemic heart disease.[21] [22] The risk was also found to be college in people with a specific genotype of the angiotensin-converting enzyme (ACE).[23]
Screening [edit]
Cholesterol screening and genetic testing among family members of people with known FH is cost-constructive.[24] Other strategies such as universal screening at the age of sixteen were suggested in 2001.[25] [26] The latter arroyo may all the same exist less cost-effective in the short term.[27] Screening at an age lower than 16 was thought probable to lead to an unacceptably loftier rate of imitation positives.[6]
A 2007 meta-analysis constitute that "the proposed strategy of screening children and parents for familial hypercholesterolaemia could have considerable impact in preventing the medical consequences of this disorder in two generations simultaneously."[28] "The use of total cholesterol lonely may best discriminate between people with and without FH between the ages of ane to 9 years."[29] [28]
Screening of toddlers has been suggested, and results of a trial on 10,000 one-year-olds were published in 2016. Piece of work was needed to find whether screening was cost-effective, and acceptable to families.[30] [31] Genetic counseling can help assist in genetic testing following a positive cholesterol screen for FH.[32]
Treatment [edit]
Heterozygous FH [edit]
FH is usually treated with statins.[ix] Statins deed by inhibiting the enzyme hydroxymethylglutaryl CoA reductase (HMG-CoA-reductase) in the liver. In response, the liver produces more LDL receptors, which remove circulating LDL from the blood. Statins effectively lower cholesterol and LDL levels, although sometimes add together-on therapy with other drugs is required, such as bile acrid sequestrants (cholestyramine or colestipol), nicotinic acid preparations or fibrates.[33] [four] Command of other risk factors for cardiovascular disease is required, as hazard remains somewhat elevated even when cholesterol levels are controlled. Professional person guidelines recommend that the decision to treat a person with FH with statins should not be based on the usual take chances prediction tools (such as those derived from the Framingham Heart Written report), as they are likely to underestimate the risk of cardiovascular illness; unlike the rest of the population, FH have had high levels of cholesterol since nascency, probably increasing their relative gamble.[34] Prior to the introduction of the statins, clofibrate (an older fibrate that often caused gallstones), probucol (especially in large xanthomas) and thyroxine were used to reduce LDL cholesterol levels.
More than controversial is the addition of ezetimibe, which inhibits cholesterol absorption in the gut. While it reduces LDL cholesterol, it does not appear to better a marker of atherosclerosis called the intima-media thickness. Whether this ways that ezetimibe is of no overall benefit in FH is unknown.[35]
There are no interventional studies that directly show mortality benefit of cholesterol lowering in FH. Rather, testify of benefit is derived from a number of trials conducted in people who have polygenic hypercholesterolemia (in which heredity plays a smaller role). Still, a 1999 observational study of a large British registry showed that bloodshed in people with FH had started to ameliorate in the early on 1990s when statins were introduced.[36]
A cohort report suggested that treatment of FH with statins leads to a 48% reduction in death from coronary heart disease to a betoken where people are no more likely to die of coronary centre disease than the general population. Withal, if the person already had coronary heart affliction the reduction was 25%. The results emphasize the importance of early identification of FH and handling with statins.[37]
Alirocumab and evolocumab, both monoclonal antibodies against PCSK9, are specifically indicated as offshoot to nutrition and maximally tolerated statin therapy for the treatment of adults with heterozygous familial hypercholesterolemia, who crave additional lowering of LDL cholesterol.[38] On 22 Dec 2021 USFDA approved new drug containing Inclisiran equally new treatment option for familial hypercholesterolemia information technology is to be given in combination with maximally tolerated statin therapy.[39]
Homozygous FH [edit]
Homozygous FH is harder to care for. The LDL (Low Density Lipoprotein) receptors are minimally functional, if at all. Only high doses of statins, often in combination with other medications, are modestly effective in improving lipid levels.[40] If medical therapy is not successful at reducing cholesterol levels, LDL apheresis may be used; this filters LDL from the bloodstream in a procedure reminiscent of dialysis.[iv] Very astringent cases may be considered for a liver transplant; this provides a liver with normally functional LDL receptors, and leads to rapid comeback of the cholesterol levels, but at the run a risk of complications from any solid organ transplant (such equally rejection, infections, or side-effects of the medication required to suppress rejection).[41] [42] Other surgical techniques include partial ileal bypass surgery, in which part of the small bowel is bypassed to subtract the absorption of nutrients and hence cholesterol, and portacaval shunt surgery, in which the portal vein is connected to the vena cava to allow blood with nutrients from the intestine to featherbed the liver.[43] [44] [45]
Lomitapide, an inhibitor of the microsomal triglyceride transfer protein,[46] was approved past the US FDA in December 2012 as an orphan drug for the treatment of homozygous familial hypercholesterolemia.[47] In January 2013, The United states FDA besides approved mipomersen, which inhibits the action of the gene apolipoprotein B, for the treatment of homozygous familial hypercholesterolemia.[48] [49] [50] Gene therapy is a possible time to come alternative.[51]
Evinacumab, monoclonal antibody inhibiting angiopoietin-similar poly peptide three, was approved in 2021 for adjunct therapy.[52]
Children [edit]
Given that FH is present from birth and atherosclerotic changes may brainstorm early in life,[53] it is sometimes necessary to care for adolescents or even teenagers with agents that were originally developed for adults. Due to rubber concerns, many physicians prefer to apply bile acrid sequestrants and fenofibrate as these are licensed in children.[54] Nevertheless, statins seem rubber and effective,[55] [56] and in older children may exist used as in adults.[6] [54]
An proficient console in 2006 advised on early combination therapy with LDL apheresis, statins, and cholesterol absorption inhibitors in children with homozygous FH at the highest risk.[57]
Epidemiology [edit]
The global prevalence of FH is approximately 10 1000000 people.[9] In about populations studied, heterozygous FH occurs in about one:250 people, but not all develop symptoms.[iii] Homozygous FH occurs in near 1:1,000,000.[four] [6]
LDLR mutations are more common in certain populations, presumably considering of a genetic phenomenon known as the founder effect—they were founded by a minor group of individuals, one or several of whom was a carrier of the mutation. The Afrikaner, French Canadians, Lebanese Christians, and Finns have high rates of specific mutations that make FH specially common in these groups. APOB mutations are more mutual in Central Europe.[4]
History [edit]
The Norwegian medico Dr Carl Müller first associated the physical signs, high cholesterol levels and autosomal dominant inheritance in 1938.[58] In the early 1970s and 1980s, the genetic cause for FH was described by Dr Joseph L. Goldstein and Dr Michael S. Brownish of Dallas, Texas. Initially, they found increased activity of HMG-CoA reductase, just studies showed that this did non explain the very abnormal cholesterol levels in people with FH.[59] The focus shifted to the binding of LDL to its receptor, and effects of impaired binding on metabolism; this proved to be the underlying mechanism for FH.[60] Later, numerous mutations in the poly peptide were directly identified by sequencing.[12] They subsequently won the 1985 Nobel Prize in Medicine for their discovery of the LDL receptor and its impact on lipoprotein metabolism.[61]
See as well [edit]
- Primary hyperlipoproteinemia
- Familial hypertriglyceridemia
- Lipoprotein lipase deficiency
- Familial apoprotein CII deficiency
- Akira Endo, discoverer of the first statin
References [edit]
- ^ Pejic RN (2014). "Familial Hypercholesterolemia". Ochsner Journal. 14 (four): 669–72. PMC4295745. PMID 25598733.
- ^ Goldberg, AC; Hopkins, PN; Toth, PP; Ballantyne, CM; Rader, DJ; Robinson, JG; Daniels, SR; Gidding, SS; de Ferranti, SD; Ito, MK; McGowan, MP; Moriarty, PM; Cromwell, WC; Ross, JL; Ziajka, PE; National Lipid Association Proficient Panel on Familial, Hypercholesterolemia. (June 2011). "Familial hypercholesterolemia: screening, diagnosis and direction of pediatric and adult patients: clinical guidance from the National Lipid Clan Expert Panel on Familial Hypercholesterolemia". Journal of Clinical Lipidology. five (3 Suppl): S1–8. doi:10.1016/j.jacl.2011.04.003. PMID 21600525.
- ^ a b c Akioyamen LE, Genest J, Shan SD, Reel RL, Albaum JM, Chu A; et al. (2017). "Estimating the prevalence of heterozygous familial hypercholesterolaemia: a systematic review and meta-analysis". BMJ Open. 7 (9): e016461. doi:10.1136/bmjopen-2017-016461. PMC5588988. PMID 28864697.
{{cite journal}}: CS1 maint: multiple names: authors listing (link) - ^ a b c d due east f g h i j 1000 l m due north o Rader DJ, Cohen J, Hobbs HH (2003). "Monogenic hypercholesterolemia: new insights in pathogenesis and treatment". J. Clin. Invest. 111 (12): 1795–803. doi:10.1172/JCI18925. PMC161432. PMID 12813012.
- ^ Tsouli SG, Kiortsis DN, Argyropoulou MI, Mikhailidis DP, Elisaf MS (2005). "Pathogenesis, detection and treatment of Achilles tendon xanthomas". Eur. J. Clin. Invest. 35 (4): 236–44. doi:10.1111/j.1365-2362.2005.01484.x. PMID 15816992. S2CID 38952905.
- ^ a b c d eastward f Durrington P (2003). "Dyslipidaemia". Lancet. 362 (9385): 717–31. doi:ten.1016/S0140-6736(03)14234-1. PMID 12957096. S2CID 208792416.
- ^ Jansen Air-conditioning, van Aalst-Cohen ES, Tanck MW, et al. (2004). "The contribution of classical risk factors to cardiovascular disease in familial hypercholesterolaemia: data in 2400 patients". J. Intern. Med. 256 (6): 482–90. doi:10.1111/j.1365-2796.2004.01405.ten. PMID 15554949. S2CID 25292760.
- ^ Williams RR, Hunt SC, Schumacher MC, et al. (1993). "Diagnosing heterozygous familial hypercholesterolemia using new practical criteria validated by molecular genetics". Am J Cardiol. ii (72): 171–76. doi:x.1016/0002-9149(93)90155-6. PMID 8328379.
- ^ a b c d due east f grand h i Repas TB, Tanner JR (February 2014). "Preventing early cardiovascular decease in patients with familial hypercholesterolemia". J Am Osteopath Assoc. 114 (2): 99–108. doi:ten.7556/jaoa.2014.023. PMID 24481802.
- ^ van Aalst-Cohen ES, Jansen Ac, Tanck MW, et al. (2006). "Diagnosing familial hypercholesterolaemia: the relevance of genetic testing". Eur. Heart J. 27 (eighteen): 2240–6. doi:ten.1093/eurheartj/ehl113. PMID 16825289.
- ^ Moghadasian MH, Salen G, Frohlich JJ, Scudamore CH (April 2002). "Cerebrotendinous xanthomatosis: a rare disease with diverse manifestations". Arch. Neurol. 59 (iv): 527–ix. doi:x.1001/archneur.59.iv.527. PMID 11939886.
- ^ a b Hobbs HH, Chocolate-brown MS, Goldstein JL (1992). "Molecular genetics of the LDLR gene in familial hypercholesterolemia". Hum. Mutat. 1 (6): 445–66. doi:10.1002/humu.1380010602. PMID 1301956. S2CID 5756814.
- ^ Vega GL, Grundy SM (1986). "In vivo evidence for reduced binding of low density lipoproteins to receptors as a cause of primary moderate hypercholesterolemia". J. Clin. Invest. 78 (5): 1410–iv. doi:10.1172/JCI112729. PMC423848. PMID 3771801.
- ^ Abifadel Chiliad, Varret M, Rabès JP, et al. (2003). "Mutations in PCSK9 crusade autosomal dominant hypercholesterolemia". Nat. Genet. 34 (2): 154–6. doi:x.1038/ng1161. PMID 12730697. S2CID 19462210.
- ^ Seidah NG, Khatib AM, Prat A (2006). "The proprotein convertases and their implication in sterol and/or lipid metabolism". Biol. Chem. 387 (7): 871–7. doi:ten.1515/BC.2006.110. PMID 16913836. S2CID 22395543.
- ^ Khachadurian AK, Uthman SM (1973). "Experiences with the homozygous cases of familial hypercholesterolemia. A report of 52 patients". Nutr Metab. 15 (i): 132–40. doi:ten.1159/000175431. PMID 4351242.
- ^ Dark-brown MS, Goldstein JL (1974). "Familial hypercholesterolemia: defective binding of lipoproteins to cultured fibroblasts associated with impaired regulation of 3-hydroxy-iii-methylglutaryl coenzyme A reductase activity". Proc. Natl. Acad. Sci. The statesA. 71 (3): 788–92. Bibcode:1974PNAS...71..788B. doi:10.1073/pnas.71.3.788. PMC388099. PMID 4362634.
- ^ Scientific Steering Commission on behalf of the Simon Broome Register Group (1991). "Risk of fatal coronary middle disease in familial hypercholesterolaemia". BMJ. 303 (6807): 893–half dozen. doi:ten.1136/bmj.303.6807.893. PMC1671226. PMID 1933004.
- ^ Sijbrands EJ, Westendorp RG, Defesche JC, de Meier PH, Smelt AH, Kastelein JJ (2001). "Mortality over two centuries in large pedigree with familial hypercholesterolaemia: family tree bloodshed study". BMJ. 322 (7293): 1019–23. doi:x.1136/bmj.322.7293.1019. PMC31037. PMID 11325764.
- ^ Jansen AC, van Aalst-Cohen ES, Tanck MW, et al. (2005). "Genetic determinants of cardiovascular illness hazard in familial hypercholesterolemia". Arterioscler. Thromb. Vasc. Biol. 25 (7): 1475–81. doi:x.1161/01.ATV.0000168909.44877.a7. PMID 15879303.
- ^ Wiklund, O.; Angelin, B.; Olofsson, S. O.; Eriksson, M.; Fager, M.; Berglund, L.; Bondjers, Yard. (Jun 1990). "Apolipoprotein(a) and ischaemic eye disease in familial hypercholesterolaemia". Lancet. 335 (8702): 1360–1363. doi:10.1016/0140-6736(ninety)91242-3. PMID 1971660. S2CID 27054208.
- ^ Seed, One thousand.; Hoppichler, F.; Reaveley, D.; Mccarthy, S.; Thompson, Chiliad. R.; Boerwinkle, Due east.; Utermann, Grand. (May 1990). "Relation of serum lipoprotein(a) concentration and apolipoprotein(a) phenotype to coronary heart disease in patients with familial hypercholesterolemia" (Free full text). The New England Journal of Medicine. 322 (21): 1494–1499. doi:10.1056/NEJM199005243222104. ISSN 0028-4793. PMID 2139920.
- ^ O'Malley JP, Maslen CL, Illingworth DR (19 May 1998). "Angiotensin-converting enzyme DD genotype and cardiovascular disease in heterozygous familial hypercholesterolemia". Apportionment. 97 (18): 1780–3. doi:10.1161/01.CIR.97.18.1780. PMID 9603531.
- ^ Besseling, J; Sjouke, B; Kastelein, JJ (Baronial 2015). "Screening and treatment of familial hypercholesterolemia - Lessons from the past and opportunities for the future (based on the Anitschkow Lecture 2014)". Atherosclerosis. 241 (2): 597–606. doi:ten.1016/j.atherosclerosis.2015.06.011. PMID 26115072.
- ^ Marks D, Wonderling D, Thorogood Grand, Lambert H, Humphries SE, Neil HA (June 2002). "Cost effectiveness analysis of different approaches of screening for familial hypercholesterolaemia". BMJ. 324 (7349): 1303. doi:10.1136/bmj.324.7349.1303. PMC113765. PMID 12039822.
- ^ Umans-Eckenhausen MA, Defesche JC, Sijbrands EJ, Scheerder RL, Kastelein JJ (January 2001). "Review of first five years of screening for familial hypercholesterolaemia in holland". Lancet. 357 (9251): 165–8. doi:x.1016/S0140-6736(00)03587-Ten. PMID 11213091. S2CID 25342898.
- ^ Marks D, Thorogood M, Neil HA, Wonderling D, Humphries SE (March 2003). "Comparing costs and benefits over a 10 year period of strategies for familial hypercholesterolaemia screening". J Public Health Med. 25 (1): 47–52. doi:10.1093/pubmed/fdg010. PMID 12669918.
- ^ a b Wald, David S; Bestwick, Jonathan P; Wald, Nicholas J (22 September 2007). "Child-parent screening for familial hypercholesterolaemia: screening strategy based on a meta-assay". BMJ. 335 (7620): 599. doi:ten.1136/bmj.39300.616076.55. PMC1989026. PMID 17855284.
- ^ Saenger, Amy K (1 August 2012). "Universal Lipid Screening in Children and Adolescents: A Baby Step toward Primordial Prevention?". Clinical Chemical science. 58 (8): 1179–1181. doi:10.1373/clinchem.2012.182287. PMID 22510399.
- ^ Caroline Parkinson (27 October 2016). "Toddlers 'should get eye risk test'". BBC News . Retrieved 27 October 2016.
- ^ Wald, David S.; Bestwick, Jonathan P.; Morris, Joan K.; Whyte, Ken; Jenkins, Lucy; Wald, Nicholas J. (2016). "Child–Parent Familial Hypercholesterolemia Screening in Primary Care". New England Journal of Medicine. 375 (17): 1628–1637. doi:ten.1056/NEJMoa1602777. ISSN 0028-4793. PMID 27783906.

- ^ "Genetic Counseling for Familial Hypercholesterolemia | CDC". world wide web.cdc.gov. 2020-04-01. Retrieved 2021-02-12 .
- ^ Nemati, Mohammad Hassan; Astaneh, Behrooz (2010). "Optimal direction of familial hypercholesterolemia: treatment and direction strategies". Vasc Wellness Take chances Manag. 6: 1079–1088. doi:x.2147/VHRM.S8283. PMC3004511. PMID 21191428.
- ^ National Institute for Health and Clinical Excellence. Clinical guideline 71: Familial hypercholesterolaemia. London, 2008.
- ^ Kastelein JJ, Akdim F, Stroes ES, et al. (Apr 2008). "Simvastatin with or without ezetimibe in familial hypercholesterolemia" (PDF). Northward. Engl. J. Med. 358 (fourteen): 1431–43. doi:10.1056/NEJMoa0800742. PMID 18376000. S2CID 8085257. Archived from the original (PDF) on 2020-06-29.
- ^ Scientific Steering Committee on behalf of the Simon Broome Annals Group (1999). "Mortality in treated heterozygous familial hypercholesterolaemia: implications for clinical management". Atherosclerosis. 142 (1): 105–12. doi:x.1016/S0021-9150(98)00200-7. PMID 9920511.
- ^ Neil A, Cooper J, Betteridge J, et al. (November 2008). "Reductions in all-crusade, cancer, and coronary mortality in statin-treated patients with heterozygous familial hypercholesterolaemia: a prospective registry study". Eur. Heart J. 29 (21): 2625–33. doi:10.1093/eurheartj/ehn422. PMC2577142. PMID 18840879.
- ^ Ito, MK; Santos, RD (16 May 2016). "PCSK9 inhibition with monoclonal antibodies-mod management of hypercholesterolemia". Periodical of Clinical Pharmacology. Online get-go (i): seven–32. doi:10.1002/jcph.766. PMC5215586. PMID 27195910.
- ^ "New Drug SiRNA approved for handling of familial hypercholesterolemia". 2021. Retrieved thirty December 2021.
- ^ Marais Advertizement, Blom DJ, Firth JC (January 2002). "Statins in homozygous familial hypercholesterolemia". Curr Atheroscler Rep. 4 (1): 19–25. doi:10.1007/s11883-002-0058-7. PMID 11772418. S2CID 8075552.
- ^ Bilheimer DW, Goldstein JL, Grundy SM, Starzl TE, Chocolate-brown MS (Dec 1984). "Liver transplantation to provide low-density-lipoprotein receptors and lower plasma cholesterol in a child with homozygous familial hypercholesterolemia". N. Engl. J. Med. 311 (26): 1658–64. doi:10.1056/NEJM198412273112603. PMC2975980. PMID 6390206.
- ^ Revell SP, Noble-Jamieson Yard, Johnston P, Rasmussen A, Jamieson North, Barnes ND (November 1995). "Liver transplantation for homozygous familial hypercholesterolaemia". Arch. Dis. Kid. 73 (5): 456–viii. doi:10.1136/adc.73.5.456. PMC1511367. PMID 8554367.
- ^ López-Santamaria M, Migliazza L, Gamez M, et al. (April 2000). "Liver transplantation in patients with homozygotic familial hypercholesterolemia previously treated by end-to-side portocaval shunt and ileal bypass". J. Pediatr. Surg. 35 (iv): 630–3. doi:10.1053/jpsu.2000.0350630. PMID 10770402.
- ^ Buchwald H, Varco RL, Boen JR, et al. (June 1998). "Effective lipid modification by partial ileal bypass reduced long-term coronary centre disease mortality and morbidity: five-year posttrial follow-up report from the POSCH. Plan on the Surgical Control of the Hyperlipidemias". Curvation. Intern. Med. 158 (11): 1253–61. doi:10.1001/archinte.158.xi.1253. PMID 9625405.
- ^ Bilheimer DW, Goldstein JL, Grundy SM, Chocolate-brown MS (December 1975). "Reduction in cholesterol and depression density lipoprotein synthesis after portacaval shunt surgery in a patient with homozygous familial hypercholesterolemia". J. Clin. Invest. 56 (6): 1420–30. doi:10.1172/JCI108223. PMC333120. PMID 172531.
- ^ Cuchel M, Bloedon LT, Szapary PO, et al. (January 2007). "Inhibition of microsomal triglyceride transfer poly peptide in familial hypercholesterolemia". Northward. Engl. J. Med. 356 (2): 148–56. doi:x.1056/NEJMoa061189. PMID 17215532.
- ^ "FDA approves new orphan drug for rare cholesterol disorder" (Press release). U.S. Nutrient and Drug Assistants. 26 December 2012. Archived from the original on 31 December 2012.
- ^ Astaneh, Behrooz; Makhdami, Nima; Astaneh, Vala; Guyatt, Gordon (July 2021). "The Result of Mipomersen in the Management of Patients with Familial Hypercholesterolemia: A Systematic Review and Meta-Assay of Clinical Trials". J Cardiovasc Dev Dis. 8 (vii): 82. doi:x.3390/jcdd8070082. PMC8304130. PMID 34357325.
- ^ Pollack, Andrew (29 January 2013). "F.D.A. Approves Genetic Drug to Care for Rare Disease". The New York Times.
- ^ "FDA approves new orphan drug Kynamro to treat inherited cholesterol disorder" (Press release). U.S. Food and Drug Administration. 29 January 2013. Archived from the original on 2 Feb 2013.
- ^ Grossman Chiliad, Rader DJ, Muller DW, et al. (November 1995). "A pilot study of ex vivo gene therapy for homozygous familial hypercholesterolaemia". Nat. Med. 1 (11): 1148–54. doi:10.1038/nm1195-1148. PMID 7584986. S2CID 3194865.
- ^ "Novel Drug Approvals for 2021". FDA.
- ^ Mabuchi H, Koizumi J, Shimizu M, Takeda R (February 1989). "Development of coronary heart illness in familial hypercholesterolemia". Circulation. 79 (2): 225–32. doi:10.1161/01.CIR.79.two.225. PMID 2914343.
- ^ a b Greene O, Durrington P (May 2004). "Clinical management of children and young adults with heterozygous familial hypercholesterolaemia in the UK". J R Soc Med. 97 (5): 226–9. doi:ten.1258/jrsm.97.5.226. PMC1079462. PMID 15121812.
- ^ Rodenburg J, Vissers MN, Wiegman A, Trip MD, Bakker Hard disk drive, Kastelein JJ (August 2004). "Familial hypercholesterolemia in children". Curr. Opin. Lipidol. xv (4): 405–xi. doi:10.1097/01.mol.0000137228.92396.f3. PMID 15243213. S2CID 38754088.
- ^ Wiegman A, Hutten BA, de Groot Due east, et al. (July 2004). "Efficacy and condom of statin therapy in children with familial hypercholesterolemia: a randomized controlled trial". JAMA. 292 (3): 331–seven. doi:10.1001/jama.292.3.331. PMID 15265847.
- ^ Kavey RE, Allada Five, Daniels SR, et al. (December 2006). "Cardiovascular hazard reduction in high-take a chance pediatric patients: a scientific argument from the American Heart Association Expert Panel on Population and Prevention Science; the Councils on Cardiovascular Affliction in the Immature, Epidemiology and Prevention, Nutrition, Physical Activity and Metabolism, High Blood Pressure Research, Cardiovascular Nursing, and the Kidney in Eye Illness; and the Interdisciplinary Working Grouping on Quality of Intendance and Outcomes Research: endorsed by the American University of Pediatrics". Apportionment. 114 (24): 2710–38. doi:ten.1161/CIRCULATIONAHA.106.179568. PMID 17130340.
- ^ Müller C (1938). "Xanthoma, hypercholesterolemia, angina pectoris". Acta Medica Scandinavica. 95 Suppl (89): 75–84. doi:10.1111/j.0954-6820.1938.tb19279.x.
- ^ Goldstein JL, Brownish MS (October 1973). "Familial hypercholesterolemia: identification of a defect in the regulation of iii-hydroxy-3-methylglutaryl coenzyme A reductase activeness associated with overproduction of cholesterol". Proc. Natl. Acad. Sci. U.S.A. lxx (10): 2804–8. Bibcode:1973PNAS...seventy.2804G. doi:10.1073/pnas.70.10.2804. PMC427113. PMID 4355366.
- ^ Brown MS, Goldstein JL (January 1976). "Receptor-mediated control of cholesterol metabolism". Science. 191 (4223): 150–4. Bibcode:1976Sci...191..150B. doi:10.1126/science.174194. PMID 174194.
- ^ Nobelprize.org. "Medicine 1985". Retrieved 2008-02-28 .
External links [edit]
- MedicinePlus: Familial Hypercholesterolemia
martinezpeartrut1958.blogspot.com
Source: https://en.wikipedia.org/wiki/Familial_hypercholesterolemia
0 Response to "Can I Have Familial Hypercholesterolemia if Neither of My Parents Have High Cholesterol"
Postar um comentário